遗传性耳聋基因治疗的进展、前景及挑战
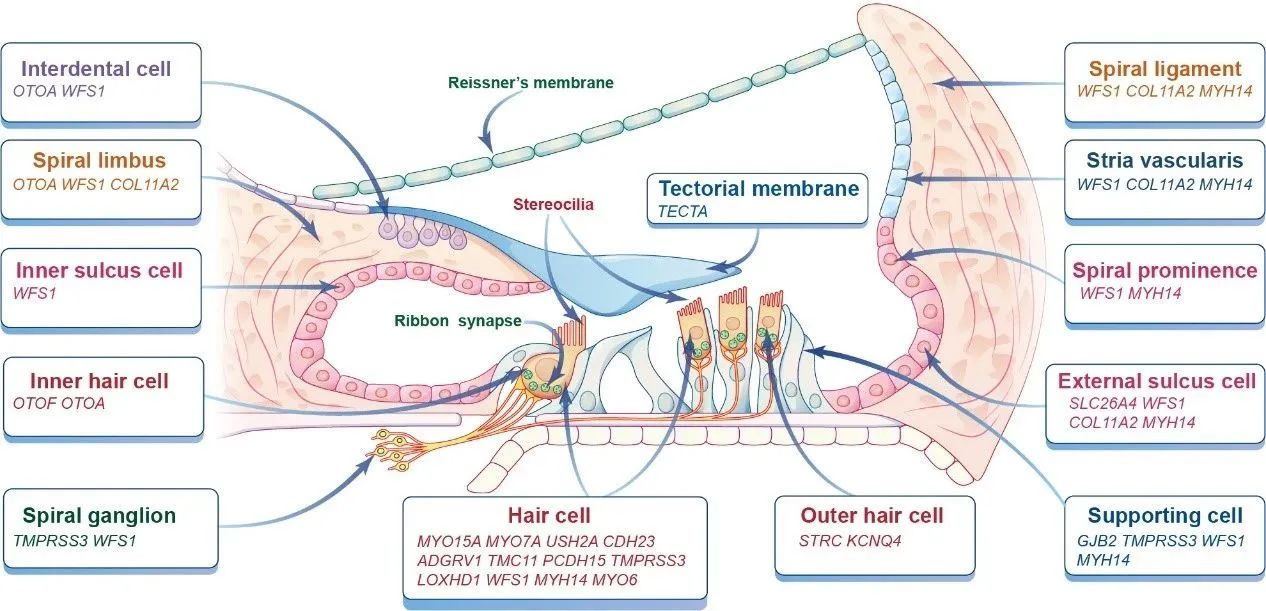
已有的研究显示,这三类致病基因是内耳基因治疗的常见靶点。当然,也有部分耳聋基因表达于耳蜗其它部位,如螺旋神经节神经元(SGN)、内外沟细胞、盖膜和Reissner膜等。通常,这些致病基因的表达部位为病毒载体的选择和治疗窗口的评估以及基因治疗系统的设计提供了有力的指导。
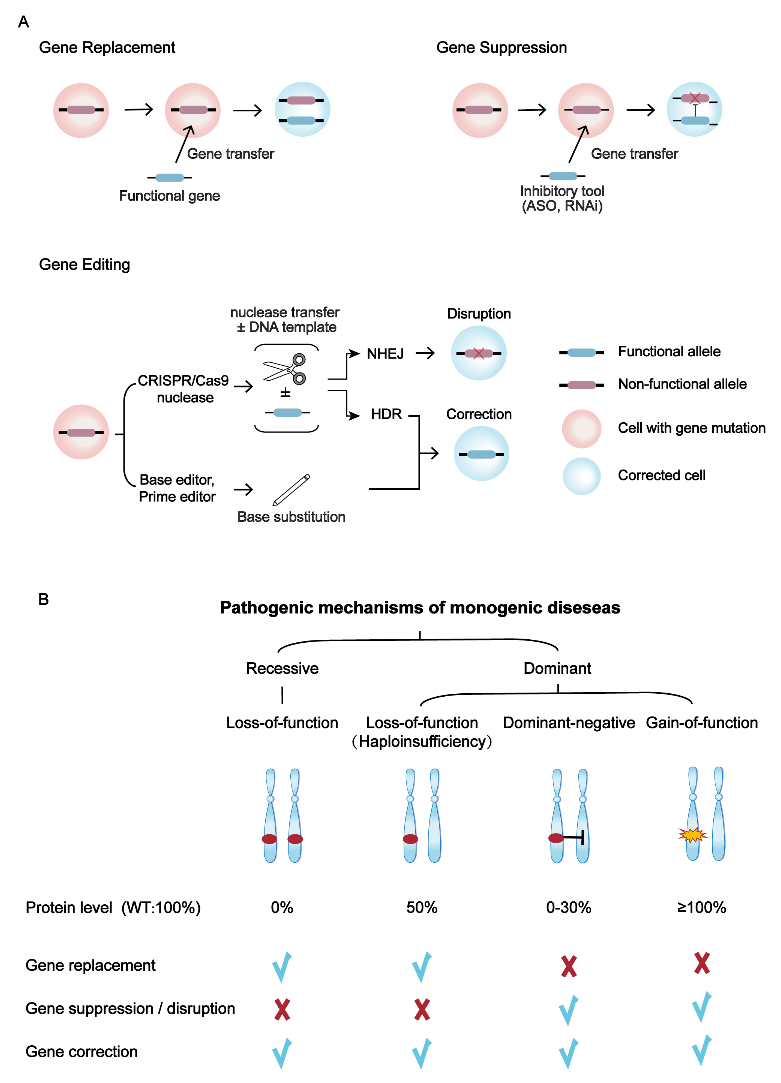
不同的基因治疗策略有着不同的工作方式(图2A)。基因替代策略是通过递送正常基因以提供充足的有功能蛋白,适用于隐性遗传病和单倍剂量不足的显性遗传病(图2)。基因抑制策略是通过反义寡核苷酸(ASO)或RNA干扰(RNAi)技术在mRNA水平抑制突变基因表达,该方法可用于DN和GOF导致的显性遗传性疾病(图2)。基因编辑策略主要包括CRISPR/Cas9核酸酶非同源末端连接(NHEJ)修复方式介导的基因敲除(gene disruption)策略,通过敲除突变基因,可应用于DN和GOF导致的显性遗传性疾病;以及CRISPR/Cas9介导的同源重组修复(HDR)、碱基编辑器和先导编辑器介导的基因修复(gene correction)策略,可直接纠正基因突变位点,因此显性、隐性遗传性疾病均适用(图2)。